分子間相互作用
分子ごとに構造を調整しつつ,分子間相互作用モデルを作成します.
これは,libbuilcule の分子構造の調整をアプリケーションレベルで実装した機能です.
分子どうしを手作業で噛み合わせるより多少まし,といった処理だと思います.
相互作用の計算には 2 種類あります.
「相互作用過程のステップ」では,分子間で静電的相互作用とファンデルワールス反発を発生させることにより,分子間相互作用モデルを作成します.
「凝集過程のステップ」ではこれに加え,系の中心に向かって分子を引き寄せる力を発生させて計算します.
両方のステップを設定した場合は,まず「凝集過程のステップ」をおこない,ついで「相互作用過程のステップ」を実行します.
系の中心に向かって分子を引き寄せる力で強制的に分子を会合させるわけですが,その力は例えばナトリウムイオンどうしなら,お互いに反発する程度の力です.
入力構造
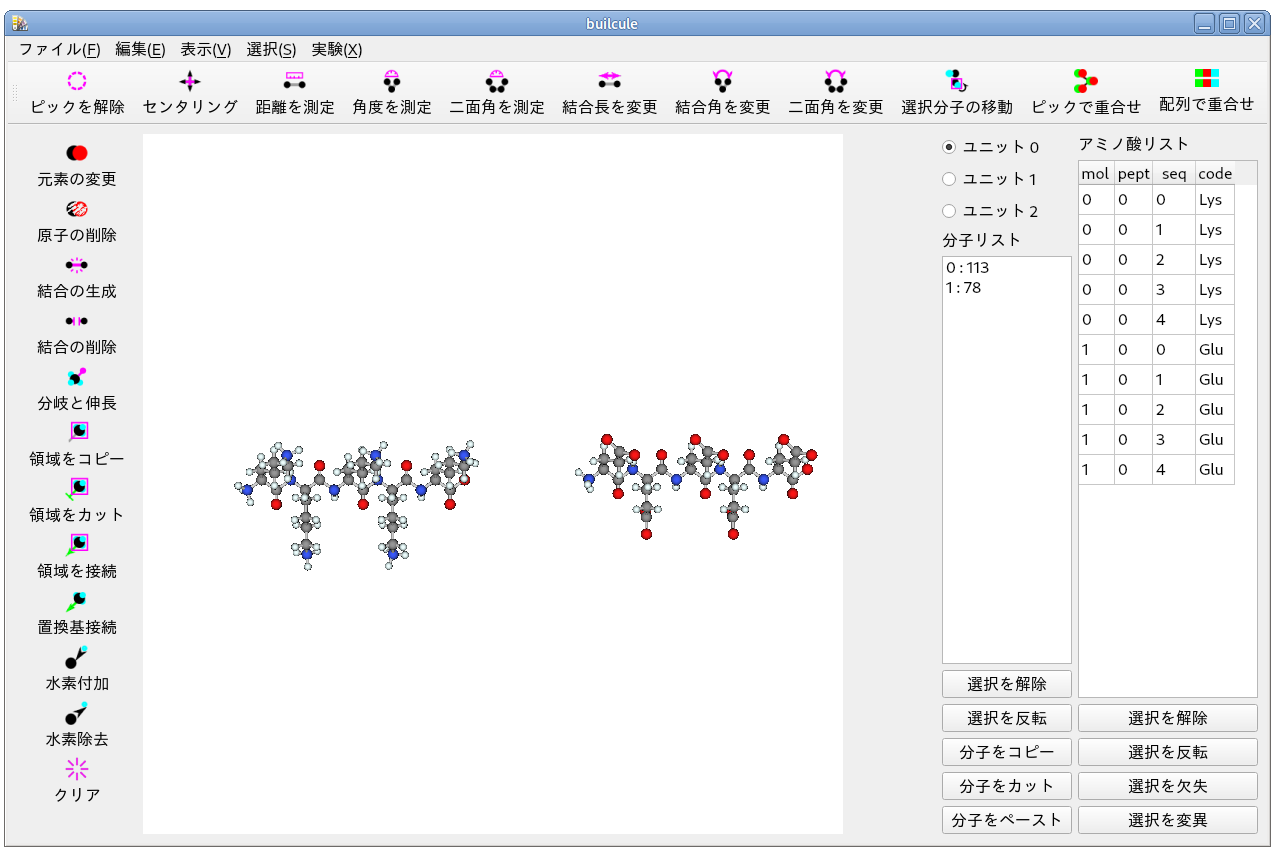
複数の分子を作成してください.
ここでは,[編集(E)]-[ペプチドを追加(P)] を使い,Lys-Lys-Lys-Lys-Lys と Glu-Glu-Glu-Glu-Glu を作成しました.
[ペプチドを追加(p)] では水素が作成されません.
これでは計算できないので,水素を付加する必要があります.
ここでは,垂直ツールバーで [水素付加] を実行しています.
分子に初期の位置を設定してください.
画像の例では [実験(X)]-[分子の位置を調整] を実行しました.
手動で分子を移動しても構いません.
計算条件
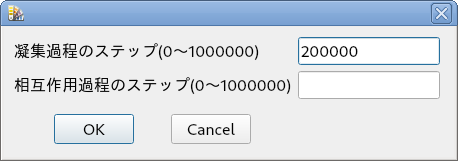
複数の分子を作成したら,[実験(X)]-[分子間相互作用] を実行してください.画像に示すダイアログボックスが表示されます.
このページの冒頭で紹介した計算のステップ数を設定してください.
画像では,凝集過程を 200000 ステップと設定しています.
数字は,ようすを見つつ,小さい値から大きい値に変更してください.
数値の設定値が大きいのは,少しずつ構造を変化させているためです.
[OK] ボタンをクリックするとシミュレーションが始まります.
計算中は一定ステップごとに再描画するので,分子が「うねうね」動くようすを目にすることができます.
凝集過程の計算結果

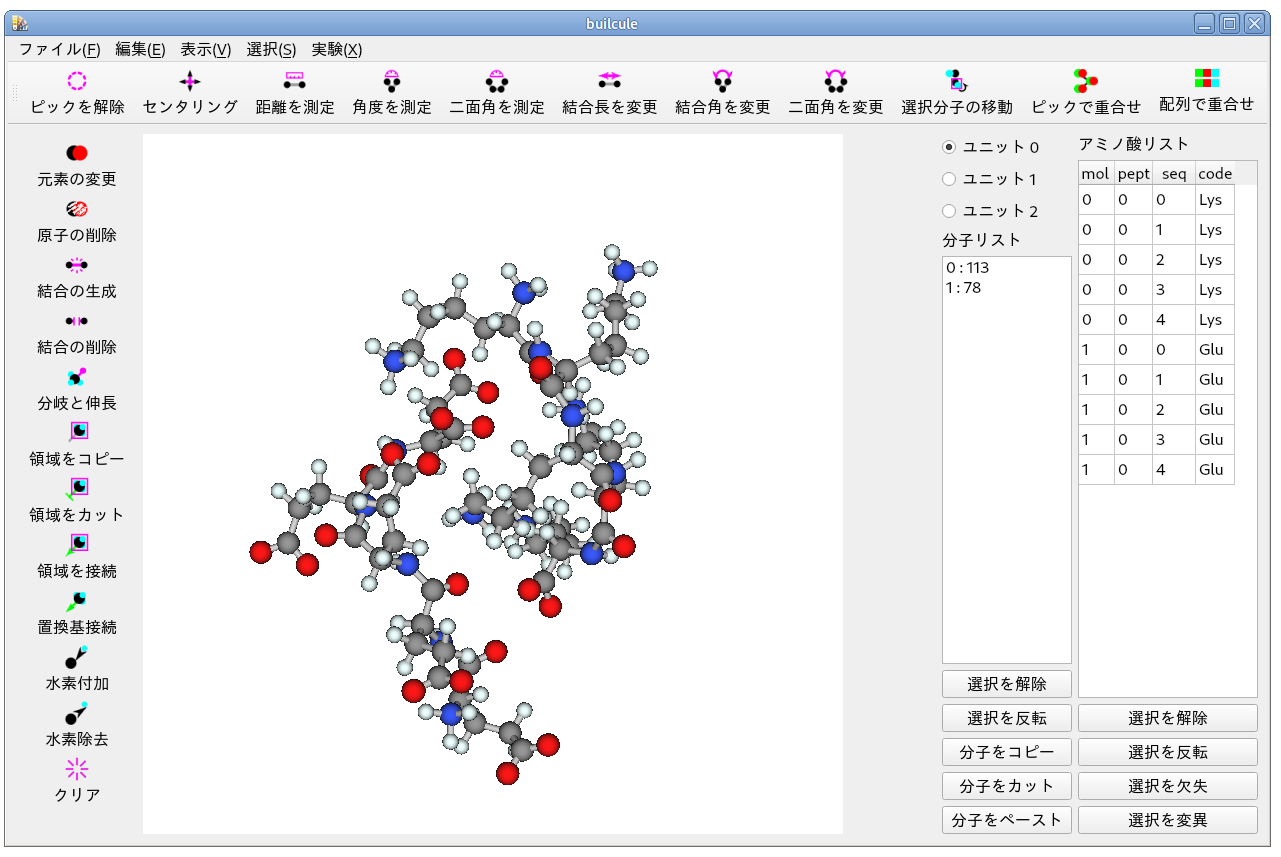
画像は凝集過程を 200000 ステップ実行したときの出力構造です.
複合体モデルが生成しているように見えます.
アミノ基とカルボキシル基のペアがいくつか形成されているように見えます.
電荷が異なるオリゴペプチドを凝集過程で処理したので,まあ,当然といえば当然なのですが.
相互作用過程の計算結果
画像は,上の複合体モデルを入力構造として相互作用過程を 100000 ステップ実行したときの出力構造です.
複合体構造が維持されているようです.
こちらの構造でも,アミノ基とカルボキシル基のペアがいくつか形成されているように見えます.