NWChem 練習ノート
まず NWChem ドキュメントの例を試し,次いでアンモニア,モノフルオロメタン,および cis-ブタジエンについて分子軌道と双極子や Mulliken 電荷など静電的な性質を計算します.
分子軌道の計算結果を MOLDEN 形式で出力すると,Gabedit で分子軌道を表示できました.
さらに,時間依存密度汎関数という法を使って,紫外-可視スペクトルを計算してみます.
このページは Debian 12 "bookworm" で作成しました.Debian 13 ”trixie” へのアップデートは,本サイトで開発している分子モデリングソフト Builcule がある程度進んでからの予定です.
インフォメーション
ソフトウェア
外部のソフトウェアは,下記バージョンの組み合わせで 分子軌道の表示に成功しました.
- Builcule:入力構造の作成(自作ソフト)
- NWChem 7.0.2
- Gabedit 2.5.1
ドキュメント
サイト:NWChem
- 実行法:[Introduction]-[Getting Started] に実行例が紹介されています
- 入力ファイル:[NWChem Examples]-[Sample Input File] には入力ファイルの例があります
- 入力ファイルは,XYZ 形式を編集すれば作成できそうです.出力ファイルは,OpenBabel で開くことも可能です
- 基底関数:Gaussian basis setsというページに一覧が載っています
- 汎関数:[Quantum Mechanics Methos]-[Density Functional Theory(DFT)] に一覧が載っています
目次(ページ内リンク)
Dunning cc-pvdz 基底系を使用した窒素分子の SCF 構造最適化(ドキュメントの計算例です)
二重項状態の水:Møller-Plesset 摂動理論(MP2)を使用した H2O+ の最適化と振動周波数(ドキュメントの計算例です)
アンモニアの分子軌道
モノフルオロメタンと cis-ブタジエンの分子軌道
紫外-可視スペクトルの計算:いくつかのアルケンの UV スペクトルを計算しました
Dunning cc-pvdz 基底系を使用した窒素分子の SCF 構造最適化
ドキュメントの最初の例を試してみます.解説部分は "Getting Started" からの改変して引用しました.詳しくはそちらを参照してください.
入力ファイル
入力ファイルを引用します.読みやすくなるかと思い,空行を追加しています.
ファイル名は n2.nw とします.ちなみに,デフォルトのファイル名は "nwchem.nw" だそうです.
入力ファイルはフリーフォーマットで,ディレクティブと呼ばれるコマンドとそのコマンドを適用するデータ(基底系,原子とその座標など)で構成されています.
この例では,TITLE,GEOMETRY,BASIS,TASK の 4 つがディレクティブです.ドキュメントの一部を引用します.
- geometry ディレクティブの n は,窒素です.データ元素などの大文字と小文字は区別されません
- geometry に引数がありませんが,デフォルト設定のカルテシアン座標と Å と解釈されます
- basis ディレクティブには,元素ごと基底系を記述します
- task ディレクティブでは,SCF エネルギーを最小化することによって分子形状を最適化するようにプログラムに対して指示しています
"Getting Started" から Geometry,Basis,Task というページへのリンクが張られており,さらに詳細な情報が得られます.
title "Nitrogen cc-pvdz SCF geometry optimization"
geometry
n 0 0 0
n 0 0 1.08
end
basis
n library cc-pvdz
end
task scf optimize
実行と結果
結果は標準出力されます.
$ nwchem n2 | tee n2.out
とすれば,結果は標準出力されつつ,file.out というファイルにも出力できます.
他に,バイナリファイルがいろいろ生成しました.
なお,コンパイルのページに並列処理のコマンドを使うことができると記してあります.すなわち,
$ mpirun -np 6 nwchem n2 | tee n2.out
として,プロセス数を指定して実行することも可能です(ここでは 6).
二重項状態の水:2 次の Møller-Plesset 摂動理論(MP2)を使用した H2O+ の最適化と振動周波数
ドキュメントの 2 番めの例です.解説部分は "Getting Started" からの改変して引用しました.詳しくはそちらを参照してください.
ドキュメントの抜粋を再編して記します.
入力ファイルには,初期値の設定とそれに続く 3 段階のタスクが記述されています.
- 初期値の入力 == ランタイムデータベースの設定
- タスク 1:計算コストの低い基底系(STO-3G)を用いて,SCF の幾何学的最適化の予備的な計算を行います
- タスク 2:MP2 と分極関数を含む基底系を使用して最適化を終了します
- タスク 3:MP2 振動周波数を計算します
まず,初期値の入力部分.
プログラムはこのデータベースから情報を取得するので,異なるブロックや指令の順番を入れ替えても,結果には影響しないそうです.
print level が low に設定されているのは,最初の計算の冗長な出力を避けるためです.
座標,電荷,基底系に続いて,scf ...end ブロックで最初の SCF 計算が設定されています.
- rhf(Unrestricted Hartree-Fock)を選択.デフォルトは rohf(restricted open-shell high-spin Hartree-Fock)
- スピン多重度は doubletを使用(デフォルトは singlet)
次いで,1 番めのタスク.
タスクでは,直前の指示文で設定されたパラメータを用いて,指定された計算をコードに実行させます.
タスクが完了しても,新しい入力ディレクティブで上書きされない限り,データベース内の設定は変更されずに後続のタスクで使用されます.
ここでは,下の内容が記述されています.
- キーワード scf と optimize で指定しているように,SCF 計算と構造最適化をおこないます
- 次のタスクのために,より適した基底系(6-31G**)を設定し直しています
次いで,2 番めのタスクでは,1 番めのタスクで設定した基底系を使って MP2 構造最適化をおこなっています.
3 番めのタスクではは振動周波数を計算しています.print 文で,SCFとMP2モジュールからの出力を無効化しています.
start h2o_freq
charge 1
geometry units angstroms
O 0.0 0.0 0.0
H 0.0 0.0 1.0
H 0.0 1.0 0.0
end
basis
H library sto-3g
O library sto-3g
end
scf
uhf; doublet
print low
end
title "H2O+ : STO-3G UHF geometry optimization"
task scf optimize
basis
H library 6-31g**
O library 6-31g**
end
title "H2O+ : 6-31g** UMP2 geometry optimization"
task mp2 optimize
mp2; print none; end
scf; print none; end
title "H2O+ : 6-31g** UMP2 frequencies"
task mp2 freq
実行と結果
入力ファイルの名前を h2o.nw とします.
拡張子を省略せずプロセス数を指定して実行し,標準出力をファイルに保存するなら,
$ mpirun -np 6 nwchem h2o.nw | tee h2o.out
とでもすれば,計算結果が h2o.out というファイルに出力できます.
アンモニアの分子軌道
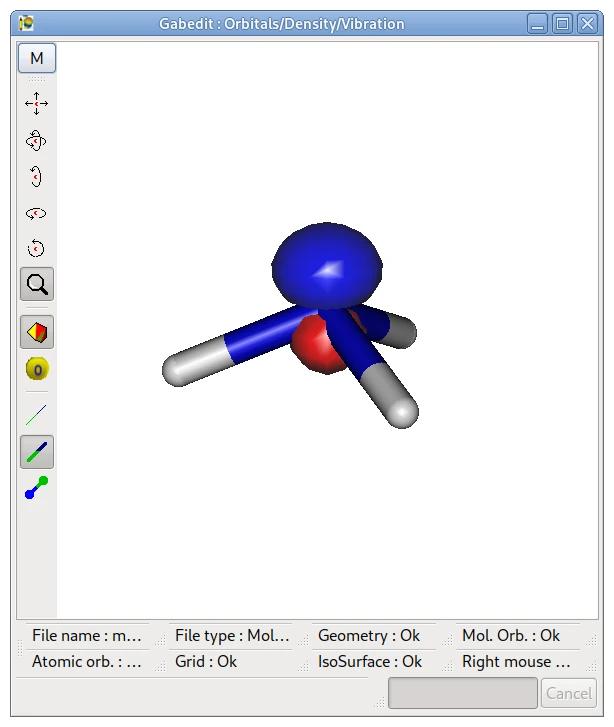
画像は,下記の計算で得たアンモニアの HOMO を Gabedit で表示したものです.
表示法は後述します
NWChem による計算法
入力ファイルの一例を下に示します.座標の部分は Builcule で作成しました.
start molecule
memory total 16 gb #メモリ
geometry
N 0.00451 0.05939 0.27871
H 1.06380 0.11517 -0.13888
H -0.41155 -0.99012 0.12048
H -0.65675 0.81556 -0.26030
end
charge 0 #電荷
basis #spherical
* library 6-31G* #基底関数セット
end
dft
mult 1 #スピン多重度
xc b3lyp #汎関数
end
property
dipole #双極子
esp #静電ポテンシャル
mulliken #MULLIKEN_電荷
#軌道を Molden 形式で出力
moldenfile
molden_norm nwchem
end
#task dft
task dft optimize #引数がない場合は一点エネルギー計算
task dft property
これを適当なディレクトリに保存して,計算を始めます.
結果は端末に標準出力されるので,パイプと tee を使って端末だけでなくファイルにも保存するのがよさそうです.
例えばファイル名を nh3.nw とし,出力したいファイル名を nh3.out すると,
$ nwchem nh3.nw | tee nh3.out
とすれば計算が始まります.計算結果は,nh3.out というファイルにも保存されます.
MOLDEN 形式の軌道は,molecule.molden というファイルに出力されます("molecule" というのは入力ファイル冒頭で設定した名前).
計算結果
最適化構造
nh3.out には構造最適化後の座標が記されています.
その値を引用します.
- N-H 結合距離(Å):1.01944
- H-N-H 結合角(°):105.76
静電的な性質
property で指定した静電的な性質を,出力ファイル nh3.out から抜粋します.
Total dipole 1.9121823905 DEBYE(S)
---------------------------------------------
Electrostatic potential/diamagnetic shielding
---------------------------------------------
1 a.u. = 9.07618 esu/cm ( or statvolts )
Point X Y Z Total Potential(a.u.) Diamagnetic shielding(a.u.)
1 N -0.00000 0.00000 0.24072 -18.377909 19.935172
2 H 0.86658 -1.54759 -0.51110 -1.066116 5.350743
3 H -1.77354 0.02331 -0.51110 -1.066116 5.350743
4 H 0.90696 1.52428 -0.51110 -1.066116 5.350743
----------------------------
Mulliken population analysis
----------------------------
Total S,P,D,... shell population
--------------------------------
Atom S P D
--------------------------------------------------------------------------------------
1 N 3.71713 4.14049 0.03004
2 H 0.70411 0.00000 0.00000
3 H 0.70411 0.00000 0.00000
4 H 0.70411 0.00000 0.00000
----- Total gross population on atoms ----
1 N 7.0 7.88766
2 H 1.0 0.70411
3 H 1.0 0.70411
4 H 1.0 0.70411
アンモニアの分子軌道
分子軌道を表示するための,Gabedit の操作法を示します.
$ gabedit &
で起動します.
- まず,分子軌道表示用ウィンドウを開きます.ツールバーにボタンがあります(マウスポインタを置くと,Display Geometry/Orbitals/Density/Vibration と表示されます)
- 分子軌道表示用ウィンドウの左上の "M" とラベルされたボタンまたは描画領域を右クリックするとポップアップメニューが表示されます
- [Orbitals]-[Read geometry and orbital from a Molden file] と進んで,上で作成した molecule.molden を開きます
- まず軌道選択用のダイアログボックスが表示されるので,目的とする軌道を選択します.デフォルトでは Alpha orbitals タブの HOMO が選択されています
- 次いで,分子軌道表示用のダイアログボックスがいくつか現れます(等高線のしきい値など).最初はデフォルト値でいいと思います
モノフルオロメタンと cis-ブタジエンの分子軌道
モノフルオロメタン
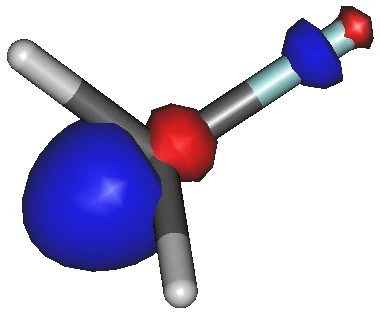
画像は,モノフルオロメタンの LUMO です.
棒様式で表示しているので,水色の棒がフッ素です.
計算法はアンモニアの場合と同じなので省略します.
SN2 反応では,
求核剤が炭素原子上に見えている青色の軌道を攻撃し,それと反結合性の赤色の軌道が切断する,
だったでしょうか.
攻撃される炭素上の LUMO(青色の軌道)の大きさに比べて.切断される結合(赤色の軌道)は相対的に小さいです,
cis-ブタジエン
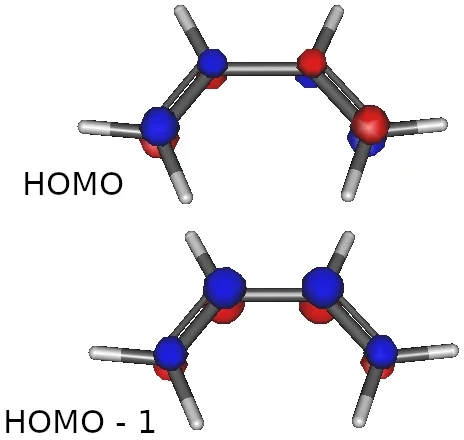
画像は,cis-ブタジエンの HOMO とそのひとつ下の軌道です.
計算法はアンモニアの場合と同じなので省略します.
こんなストーリーだったでしょうか.
(1) ブタジエンには,π電子が4個あって2個の分子軌道を作成する
(2) この場合,エネルギー準位の高い方の軌道が HOMO
(3) エネルギー準位の低い軌道が,電子がより非局在化している
計算して軌道を描いたら,図のように教科書で見たような形になりました.
紫外-可視 スペクトルの計算
時間依存密度汎関数 (TD-DFT) 法を使うと,紫外-可視 (UV) スペクトルを計算できます.
NWChem を使う計算法とその具体例が,CIS, TDHF, TDDFT で紹介されています.さらに,計算の出力からスペクトルを抽出する nw_spectrum.py という Python スクリプトもダウンロードできます.
ここでは,UV スペクトルの吸収極大が容易に入手できる,エチレンおよびいくつかの共役ジエンについて,練習してみました.NWChem による計算は一点計算です.
計算例:エチレンおよびいくつかの共役ジエンの UV スペクトル
NWChem での計算
下に,エチレンの場合の入力ファイルの内容を示す.「CIS, TDHF, TDDFT」に記載されている入力ファイルを参考にして作成しました.
START molecule #分子の名前.中間ファイルの名前として使われる GEOMETRY C -0.57187 -0.29348 0.18008 H -1.49746 -0.08238 -0.34660 C 0.57287 0.29291 -0.17902 H 1.49943 0.08374 0.34557 H -0.60764 -0.99604 1.00622 H 0.60467 0.99525 -1.00626 END charge 0 #エチレンの場合省略してもいいが,アントシアン類では必要になる場合がある BASIS * library cc-pvdz #基底関数群 END DFT xc b3lyp #汎関数 END TDDFT cis #Tamm-Dancoff 近似を使う nroots 20 #励起状態の数.実際より数個多い値を設定する.デフォルトは 1 end TASK tddft energy
入力ファイル名を input.nw とします.
計算の経過と結果は標準エラー出力されるので,私は下のように tee コマンドをパイプしてファイルにリダイレクトしています.下の例では,出力ファイルは output.nw となります.
$ wchem input.nw | tee output.nw
スペクトルデータの抽出
ダウンロードした nw_spectrum.py を使ってみます.今度は output.nw が入力ファイルとなります.
nw_spectrum.py および output.nw を同じディレクトリに置いて,そのディレクトリで,
$ python3 nw_spectrum.py -wnm < output.nw > spectrum.dat
とすると,spectrum.dat というファイルにスペクトルデータが抽出されます.ここでは,オプション引数はすべてデフォルトとしています.
spectrum.dat の冒頭を下に示します.196 nm からの吸光度が記されました.
抽出されたスペクトルは,波長と吸収を記述した 2 列のテキストファイルです.
これをデータとして折れ線グラフを描けば,UV/Vis スペクトルとなります.ここでは,Python / Pandas で描きます.
# ================================
# NWChem spectrum parser ver 2.2
# ================================
#
# Parser runtime options: {'infile': None, 'outfile': None, 'datafmt': 'auto', 'width': 0.1, 'nbin': 20, 'npoints': 2000, 'units': 'nm', 'delim': ' ', 'makespec': True, 'header': True, 'cchar': '#', 'verbose': False}
#
# The input appears to contain TDDFT data.
# Successfully parsed 20 TDDFT singlet excitations
# Roots were Lorentzian broadened with width 0.1 eV
# Spectrum generated with 2000 points.
#
# Wavelen. [nm] Abs. [au]
#----------------------------------
1.9699675854e+02 0.0000000000e+00
1.9682841593e+02 0.0000000000e+00
1.9666036080e+02 0.0000000000e+00
1.9649259239e+02 0.0000000000e+00
1.9632510999e+02 0.0000000000e+00
1.9615791285e+02 0.0000000000e+00
1.9599100025e+02 3.4889977994e-07
1.9582437146e+02 3.5078910876e-07
1.9565802577e+02 3.5269382235e-07
1.9549196244e+02 3.5461408815e-07
1.9532618077e+02 3.5655007591e-07
吸収極大波長
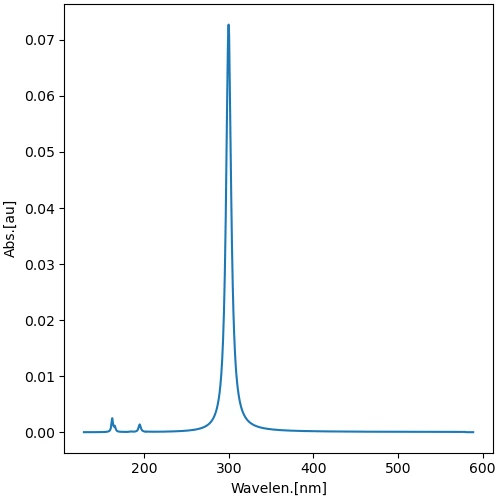
いくつかの化合物について吸収極大波長を求め,一覧表にまとめました.実験値はウェブ検索で得た値です.
エチレン以外は,2〜3% 程度の誤差で吸収極大波長が得られまし.
画像は,1,3,5,7-オクタテトラエンのスペクトルデータから作成した UV/Vis スペクトルです.
| 化合物名 | LUMO | HOMO | LUMO - HOMO | 計算値(nm) | 実験値(nm) |
|---|---|---|---|---|---|
| エチレン | 0.007179 | -0.273121 | 0.280300 | 141 | 162 |
| 1,3-ブタジエン | -0.055247 | -0.222878 | 0.167631 | 212 | 217 |
| 1,3,5−ヘキサトリエン | -0.076342 | -0.204870 | 0.128528 | 259 | 266 |
| 1,3,5,7-オクタテトラエン | -0.088113 | -0.193884 | 0.105771 | 300 | 303 |
| 1,3,5,7,9-デカペンタエン | -0.096945 | -0.186305 | 0.0893600 | 340 | 334 |
計算結果は,以下のようにまとめられそうです.
- 共役系が 1 個増えると,吸収極大波長が長波長側に 40〜50 nm シフトしている
- シフトの大きさは,共役ジエンが長くなるほど小さくなる
- 共役系が伸びるにしたがい,LUMO - HOMO は小さくなっている
発色に伴う励起が HOMO から LUMO への電子の移動に相当するかどうか知らないのですが,共役系が長いほど励起エネルギーが小さいという結果になりました.