PSI4 練習ノート
まずドキュメントの最初の例(水の計算)を試します.
それを引き継いでもう少し水の計算をおこない,さらにモノクロロメタン,および trans-ブタジエンについて分子軌道と双極子や Mulliken 電荷など静電的な性質を計算します.
分子軌道の計算結果を MOLDEN 形式で出力すると,Gabedit で分子軌道を表示できました.
このページは Debian 12 "bookworm" で作成しました.Debian 13 ”trixie” へのアップデートは,本サイトで開発している分子モデリングソフト Builcule がある程度進んでからの予定です.
インフォメーション
ドキュメント
ドキュメントによると,PSI4 は計算負荷の高い部分が C++ で書かれ、C++ クラスのエクスポートやインターフェース部分は Python で書かれています.
PSI4 は,実行ファイルとしても Python モジュールとしても使えます.
このページでは実行ファイルとして使っています.
サイト:PsiCode
- 実行法:マニュアルの [A PSI4 Tutorial]-[Psithon Tutorial: Using PSI4 as an Executable] に記されています
- 入力ファイル:マニュアルの最初の項目が Basic Input File Structure です
- 入力ファイルは,XYZ 形式を編集すれば作成できそうです
- 基底関数:マニュアルの [Appendices]-[Basis Sets] と進むと一覧があります
- 汎関数:マニュアルの [Appendices]-[Miscellaneous]-[DFT Functionals] と進むと一覧があります
ソフトウェア
外部のソフトウェアは,下記バージョンの組み合わせで 分子軌道の表示に成功しました.
- Builcule:入力構造の作成(自作ソフト)
- PSI4: Open-Source Quantum Chemistry ドキュメントのページです
- Gabedit 2.5.1
目次(ページ内リンク)
cc-pVDZ 基底系を使った,水分子の Hartree–Fock SCF 計算(ドキュメントの計算例です)
もう少し水の計算
モノクロロメタンおよび trans-ブタジエンの分子軌道
cc-pVDZ 基底系を使った,水分子の Hartree–Fock SCF 計算
入力ファイル
下は,"Sample Input Files" の最初のものです.
ファイル名をデフォルトの input.dat として保存します.
# Any line starting with the # character is a comment line
#! Sample HF/cc-pVDZ H2O computation
memory 600 mb
molecule h2o {
O
H 1 0.96
H 1 0.96 2 104.5
}
set basis cc-pVDZ
energy('scf')
チュートリアルには,
"The program correctly guessed all of these options for us. We can change the default behavior through additional keywords."
と書いてあります.
この例から思うに,特に設定しなければ,電荷 0 の三重項状態と解釈されるようです.
実行と結果
$ si4 input.dat output.dat(このようにデフォルトのファイル名を使っている場合は,単に "psi4" だけでも可)
とすれば,結果は output.dat というファイルに出力されます.
電荷,スピン多重度,分子軌道の計算法の設定
水の例の次に,メチレンビラジカルを使い,標記の設定法が記されています.
操作は水の場合と同じなので,入力ファイルのみ引用します.行の途中にコメントを付けてみました.
- "0 3" と記してある行で電荷とスピン多重度を設定しています
- "set reference uhf" という行で,Unrestricted Hartree–Fock 計算を設定しています
#! Sample UHF/6-31G** CH2 computation
molecule ch2 {
0 3 #電荷とスピン多重度
C
H 1 R
H 1 R 2 A
R = 1.075
A = 133.93
}
set basis 6-31G**
set reference uhf #分子軌道の計算法
energy ('scf')
もう少し水の計算
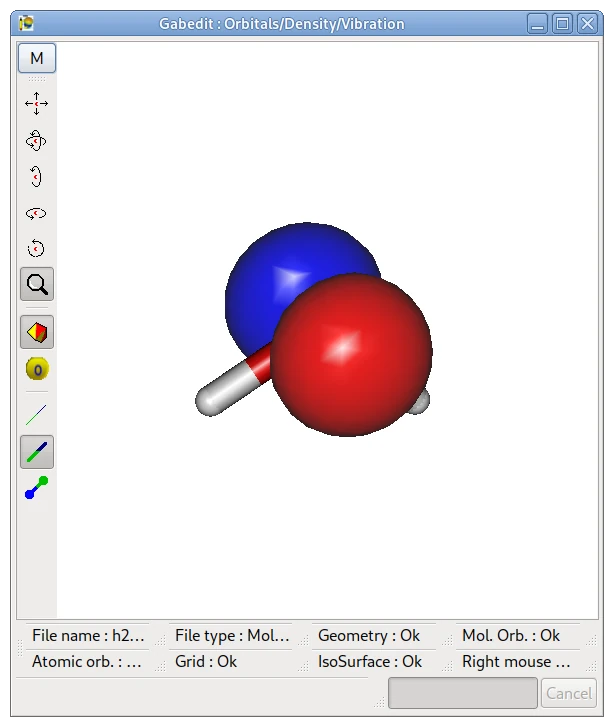
画像は,下記の計算で得た水の HOMO を Gabedit で表示したものです.
表示法は後述します
PSI4 による計算法
入力ファイルの一例を下に示します.座標の部分は Builcule で作成しました.
set_num_threads(nthread = 12) #スレッド数
set_memory("16GB") #メモリ
set {
basis = 6-31G* #基底関数セット
print 0
}
molecule h2o {
0 1 #電荷とスピン多重度
O 0.00000 0.00000 0.00000
H 0.00000 -0.65818 -0.93081
H -0.93081 0.65818 0.00000
}
scf_e, scf_wfn = optimize('b3lyp', return_wfn = True) #汎関数
oeprop(scf_wfn, 'DIPOLE') #双極子
oeprop(scf_wfn, 'ESP_AT_NUCLEI') #静電ポテンシャル
oeprop(scf_wfn, 'MULLIKEN_CHARGES') #MULLIKEN_電荷
oeprop(scf_wfn, 'LOWDIN_CHARGES') #LOWDIN_電荷
molden(scf_wfn, 'h2o.molden') #軌道を Molden 形式で出力.ファイル名は "h2o.molden" となる
これを input.dat という名前で適当なディレクトリに保存して,
$ psi4
とすれば計算が始まります.
input.dat というのはデフォルトのファイル名で,引数として省略できます.
計算結果は,output.dat というファイルに出力されます.
MOLDEN 形式の軌道は,h2o.molden というファイルに出力されます.
最適化構造
output.dat には構造最適化後の座標が記されています.
その値を引用します.
- O-H 結合距離(Å):0.968615
- H-O-H 結合角(°):103.662723
静電的な性質
oeprop で指定した静電的な性質を,出力ファイル output.dat から抜粋します.
Dipole Moment: [D]
X: 0.0000 Y: -0.0000 Z: -2.0952 Total: 2.0952
Electrostatic potentials at the nuclear coordinates:
---------------------------------------------
Center Electrostatic Potential (a.u.)
---------------------------------------------
1 O -22.322095502523
2 H -1.001829313335
3 H -1.001829313335
---------------------------------------------
Mulliken Charges: (a.u.)
Center Symbol Alpha Beta Spin Total
1 O 4.38717 4.38717 0.00000 -0.77433
2 H 0.30642 0.30642 0.00000 0.38717
3 H 0.30642 0.30642 0.00000 0.38717
Total alpha = 5.00000, Total beta = 5.00000, Total charge = -0.00000
Lowdin Charges: (a.u.)
Center Symbol Alpha Beta Spin Total
1 O 4.33672 4.33672 0.00000 -0.67344
2 H 0.33164 0.33164 0.00000 0.33672
3 H 0.33164 0.33164 0.00000 0.33672
Total alpha = 5.00000, Total beta = 5.00000, Total charge = -0.00000
水の分子軌道
分子軌道を表示するための,Gabedit の操作法を示します.
$ gabedit &
で起動します.
- まず,分子軌道表示用ウィンドウを開きます.ツールバーにボタンがあります(マウスポインタを置くと,Display Geometry/Orbitals/Density/Vibration と表示されます)
- 分子軌道表示用ウィンドウの左上の "M" とラベルされたボタンまたは描画領域を右クリックするとポップアップメニューが表示されます
- [Orbitals]-[Read geometry and orbital from a Molden file] と進んで,上で作成した h2o.molden を開きます
- まず軌道選択用のダイアログボックスが表示されるので,目的とする軌道を選択します.デフォルトでは Alpha orbitals タブの HOMO が選択されています
- 次いで,分子軌道表示用のダイアログボックスがいくつか現れます(等高線のしきい値など).最初はデフォルト値でいいと思います
モノクロロメタンおよび trans-ブタジエンの分子軌道
モノクロロメタン
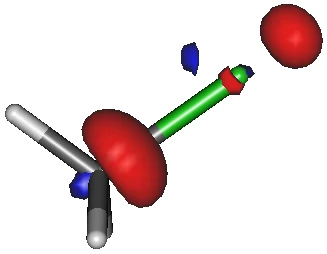
画像は,モノクロロメタンの LUMO です.
棒様式で表示しているので,緑色の棒が塩素です.
計算法は水の場合と同じなので省略します.
SN2 反応では,
求核剤が炭素原子上に見えている青色の軌道を攻撃し,それと反結合性の赤色の軌道が切断する,
だったでしょうか.
切断される結合(赤色の軌道)の大きさに比べて,攻撃される炭素上の LUMO(青色の軌道)は相対的に小さいです.
trans-ブタジエン
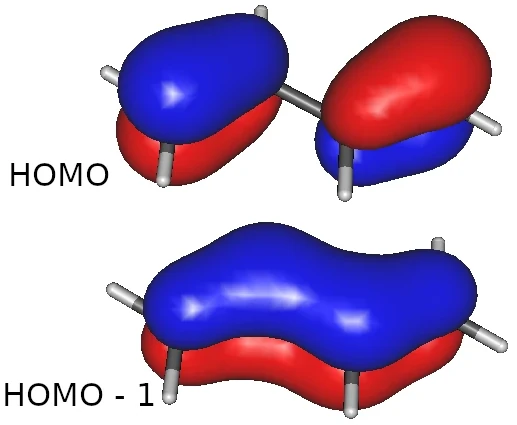
画像は,trans-ブタジエンの HOMO とそのひとつ下の軌道です.
計算法は水の場合と同じなので省略します.
こんなストーリーだったでしょうか.
(1) ブタジエンには,π電子が4個あって2個の分子軌道を作成する
(2) この場合,エネルギー準位の高い方の軌道が HOMO
(3) エネルギー準位の低い軌道が,電子がより非局在化している
計算して軌道を描いたら,図のように教科書で見たような形になりました.